Paper Sharing
【Domestic Papers】First-principle investigation of Co, N, and Co-N co-doping effects on electronic and optical properties of β-Ga₂O₃
日期:2026-05-12阅读:46
Researchers from the Hebei University of Engineering, Xi’an Rare Metal Material Institute Co. Ltd, Hebei Vocational University of Technology and Engineering, Xingtai University, Yanshan University have published a dissertation titled "First-principle investigation of Co, N, and Co-N co-doping effects on electronic and optical properties of β‑Ga₂O₃" in Journal of Physics and Chemistry of Solids.
Background
β-Ga₂O₃ is an excellent ultra-wide bandgap semiconductor, but its low intrinsic conductivity limits device applications. Doping is an effective way to tune its optoelectronic properties. However, systematic theoretical studies on Co single doping and Co‑N co-doping in β-Ga₂O₃ remain scarce. This work employs first-principles calculations to investigate the effects of Co, N single doping and Co‑N co-doping on the electronic structure, bandgap, and optical properties, providing theoretical support for experimental performance optimization.
Abstract
Ga₂O₃ is a wide-bandgap semiconductor, which has five different phases. Among these phases, β‑Ga₂O₃ is the most stable one. It has many outstanding characteristics compared to other semiconductor materials. To investigate the influence of doping on its electronic and optical properties, and given the current scarcity of studies on the effects of Co single doping and Co‑N co-doping on the optoelectronic properties of β‑Ga₂O₃ first-principles calculations were employed to investigate the effects of Co and N single doping, as well as Co‑N co-doping on the optoelectronic properties of β‑Ga₂O₃. The geometric structure, electronic structure, and optical properties of the systems were calculated and analyzed. The results show that both Co single doping and Co‑N co-doping models are thermodynamically stable structures. Electronic structure analysis reveals bandgap reduction in all doped systems relative to intrinsic β‑Ga₂O₃. Particularly, Co‑N co-doping synergistically integrates the advantages of both Co and N single doping, achieving further bandgap narrowing beyond that attained by individual Co or N doping. Optical property calculations demonstrate that Co‑N co-doping is capable of effectively facilitating the red shift in the optical absorption coefficient, exhibiting a larger optical absorption range and intensity. The research results can provide references for experimental researchers in designing β‑Ga₂O₃ with enhanced optical performance and developing efficient photocatalysts.
Highlights
This work carries out the first systematic first-principles comparative study on Co, N single-doped and Co‑N co-doped β‑Ga₂O₃, constructing 12 structural models and analyzing their stability and optoelectronic properties.
It confirms that Co‑N co-doping can synergistically narrow the bandgap, introduce impurity levels and intermediate bands, combining p-type doping characteristics and spin polarization properties.
It reveals that Co‑N co-doping significantly broadens the optical absorption range, enhances absorption intensity and induces obvious redshift, providing an optimization path for photocatalytic and photovoltaic applications.
It clarifies that Co preferentially substitutes Ga₂ site and N preferentially substitutes O₃ site; co-doping reduces formation energy and improves thermodynamic stability of the structure.
Conclusion
First-principles calculation methods were employed to study the effects of Co and N single doping, as well as of Co‑N co-doping on the optoelectronic properties of β‑Ga₂O₃ semiconductor materials. Twelve different models were studied, including intrinsic β‑Ga₂O₃ and doping systems. The results show.
(1) Most of the doped configurations exhibit thermodynamic stability. Particularly, Co more likely substitutes Ga₂. In Co‑N co-doping, Co substituting Ga₂ exhibits better thermodynamic stability compared to Co substituting Ga₁.
(2) In all doped systems, the band gap is reduced. N single doping causes the valence band maximum to intersect the Fermi level, indicating that N single doping makes β‑Ga₂O₃ metallic and belongs to p-type doping. For Co single doping, intermediate bandgaps are introduced and induces spin splitting of energy bands. This means Co single doped β‑Ga₂O₃ has potential as a semiconductor material for solar cells and spintronic devices. Particularly, Co‑N co-doping combines the advantages of impurity energy levels formed at the Fermi level in N single doping and the intermediate bands introduced by Co single doping, further reducing the bandgap of β‑Ga₂O₃.
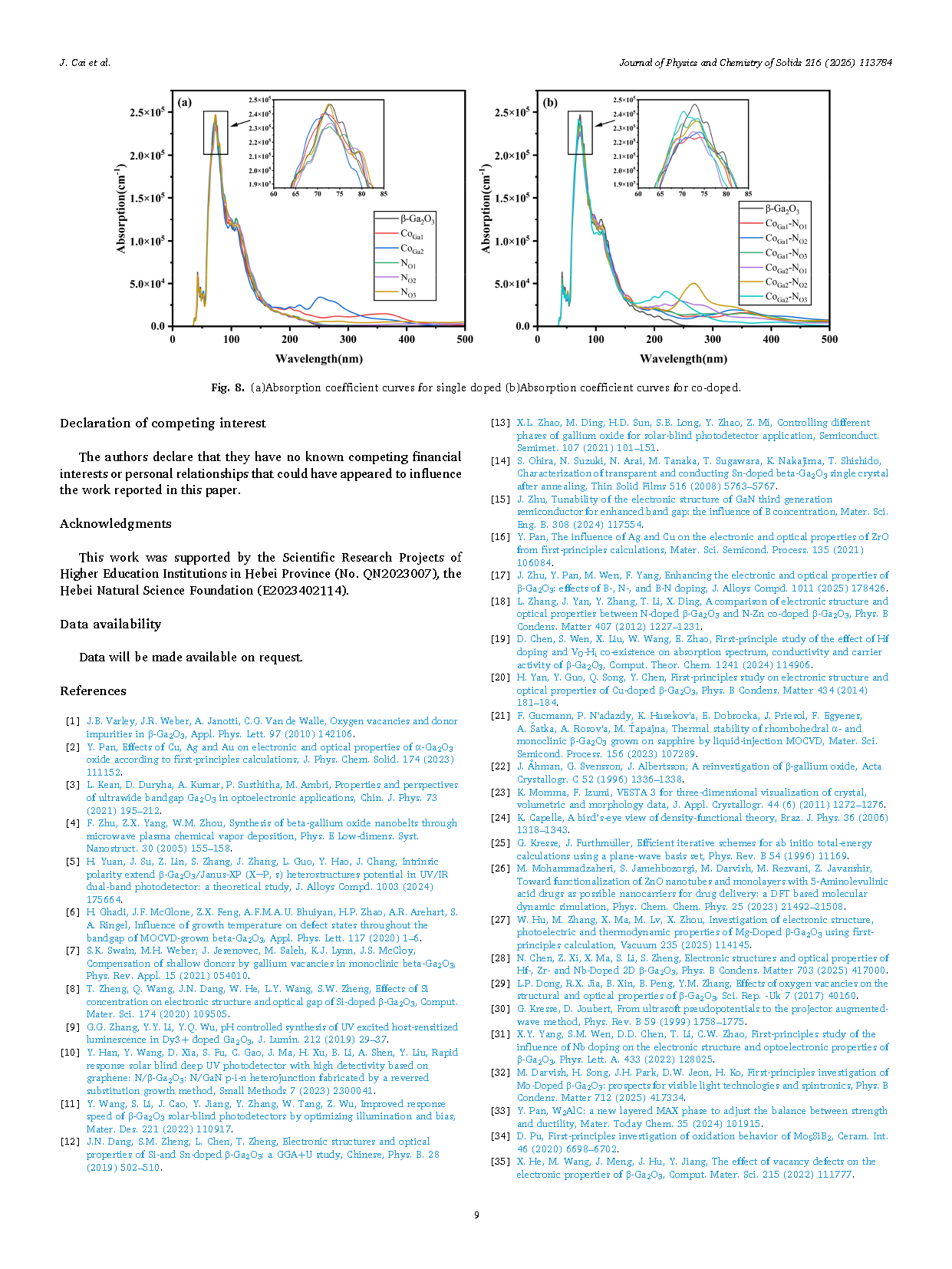
(3) Optical performance analysis results show that Co‑N co-doping can effectively promote red-shift, with a broader optical absorption range and greater intensity.
Project Support
This work was supported by the Scientific Research Projects of Higher Education Institutions in Hebei Province (No. QN2023007), the Hebei Natural Science Foundation (E2023402114).
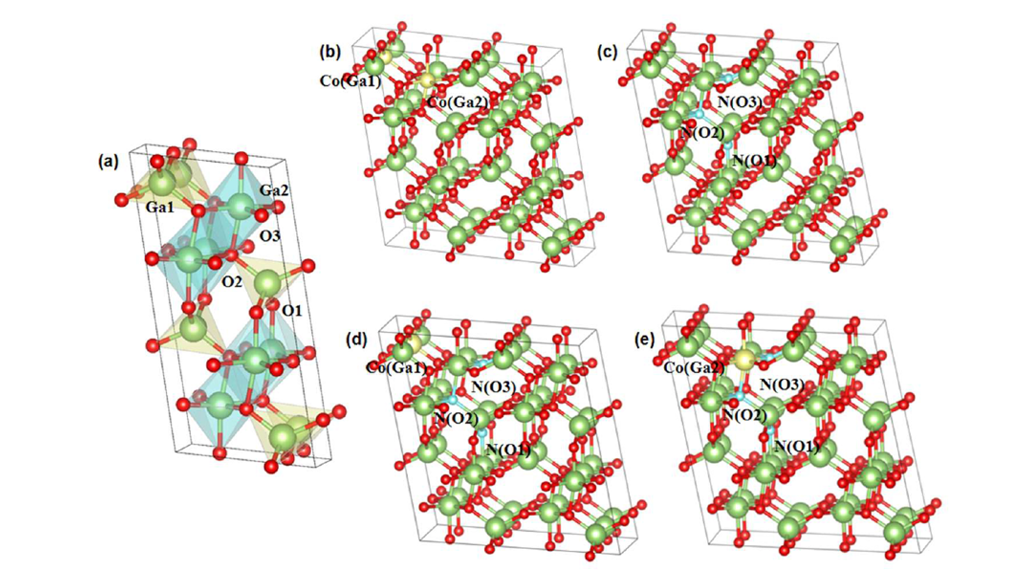
Figure 1 Structural models of doped systems: (a) intrinsic β-Ga₂O₃, (b) 1×2×2 β-Ga₂O₃ with Co doping at Ga sites, (c) 1×2×2 β-Ga₂O₃ with N doping at O sites, (d) 1×2×2 β-Ga₂O₃ with CoGa1-NO1,CoGa1-NO2, and CoGa1-NO3, (e) 1×2×2 β-Ga₂O₃ with CoGa2-NO1,CoGa2-NO2, and CoGa2-NO3
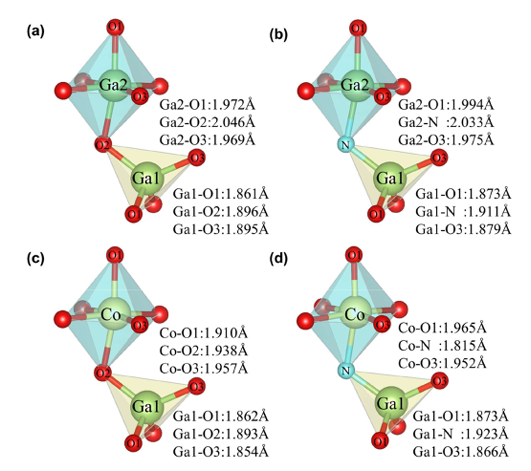
Figure 2 Partial bond lengths after structural optimization: (a) intrinsic β-Ga₂O₃ (b) β-Ga₂O₃ with N doping at O2 site, (c) β-Ga₂O₃ with Co doping at Ga2 site, (d) β-Ga₂O₃ with CoGa2-NO2
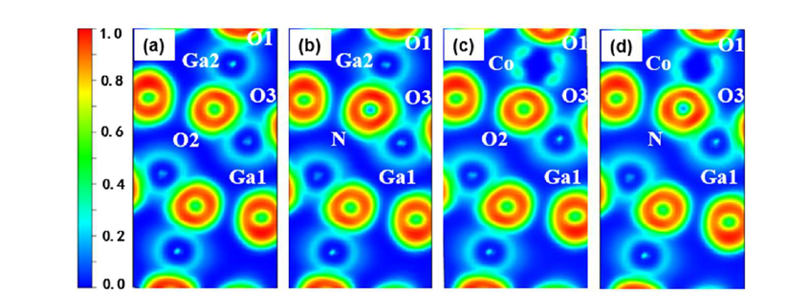
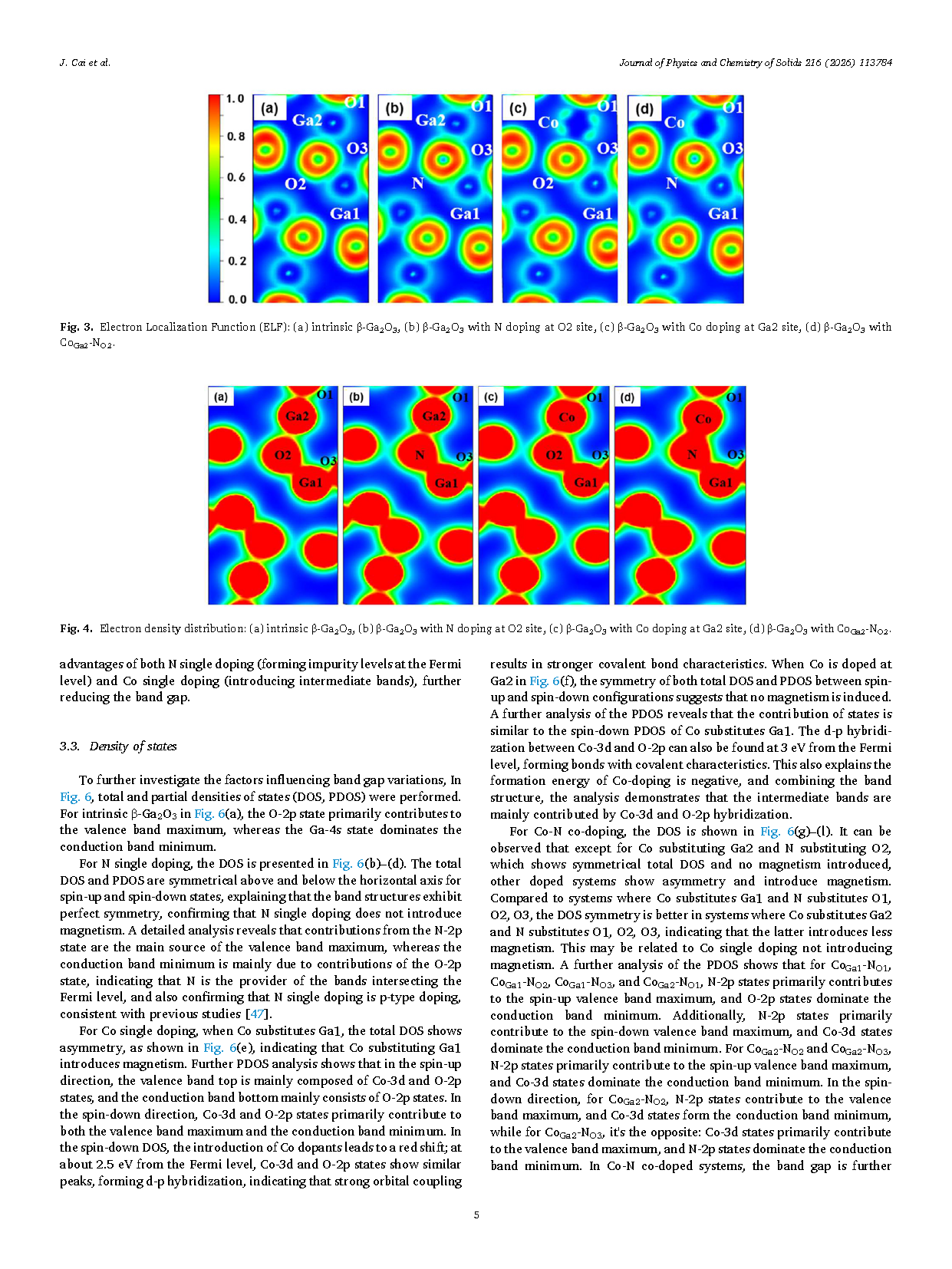
Figure 3 Electron Localization Function (ELF): (a) intrinsic β-Ga₂O₃, (b) β-Ga₂O₃ with N doping at O2 site, (c) β-Ga₂O₃ with Co doping at Ga2 site, (d) β-Ga₂O₃ with CoGa2-NO2
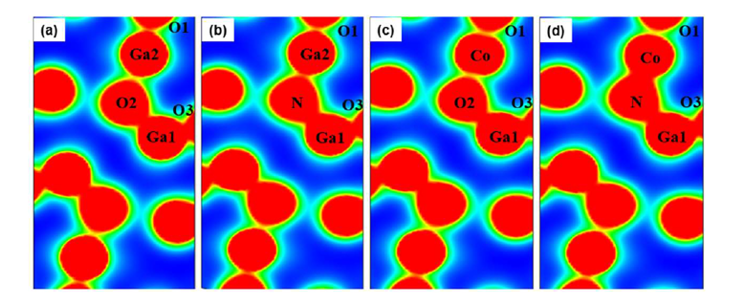
Figure 4 Electron density distribution: (a) intrinsic β-Ga₂O₃ (b) β-Ga₂O₃ with N doping at O2 site, (c) β-Ga₂O₃ with Co doping at Ga2 site, (d) β-Ga₂O₃ with CoGa2-NO2
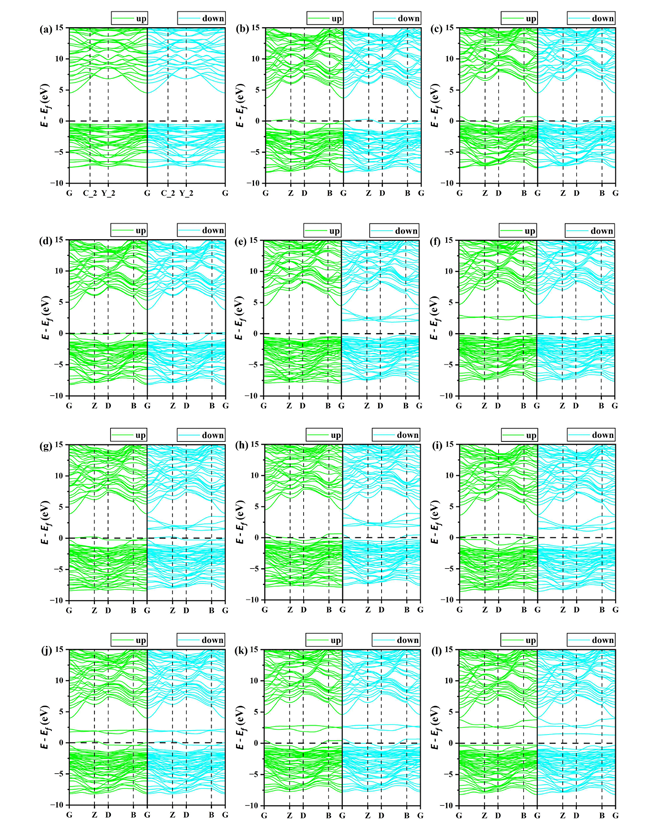
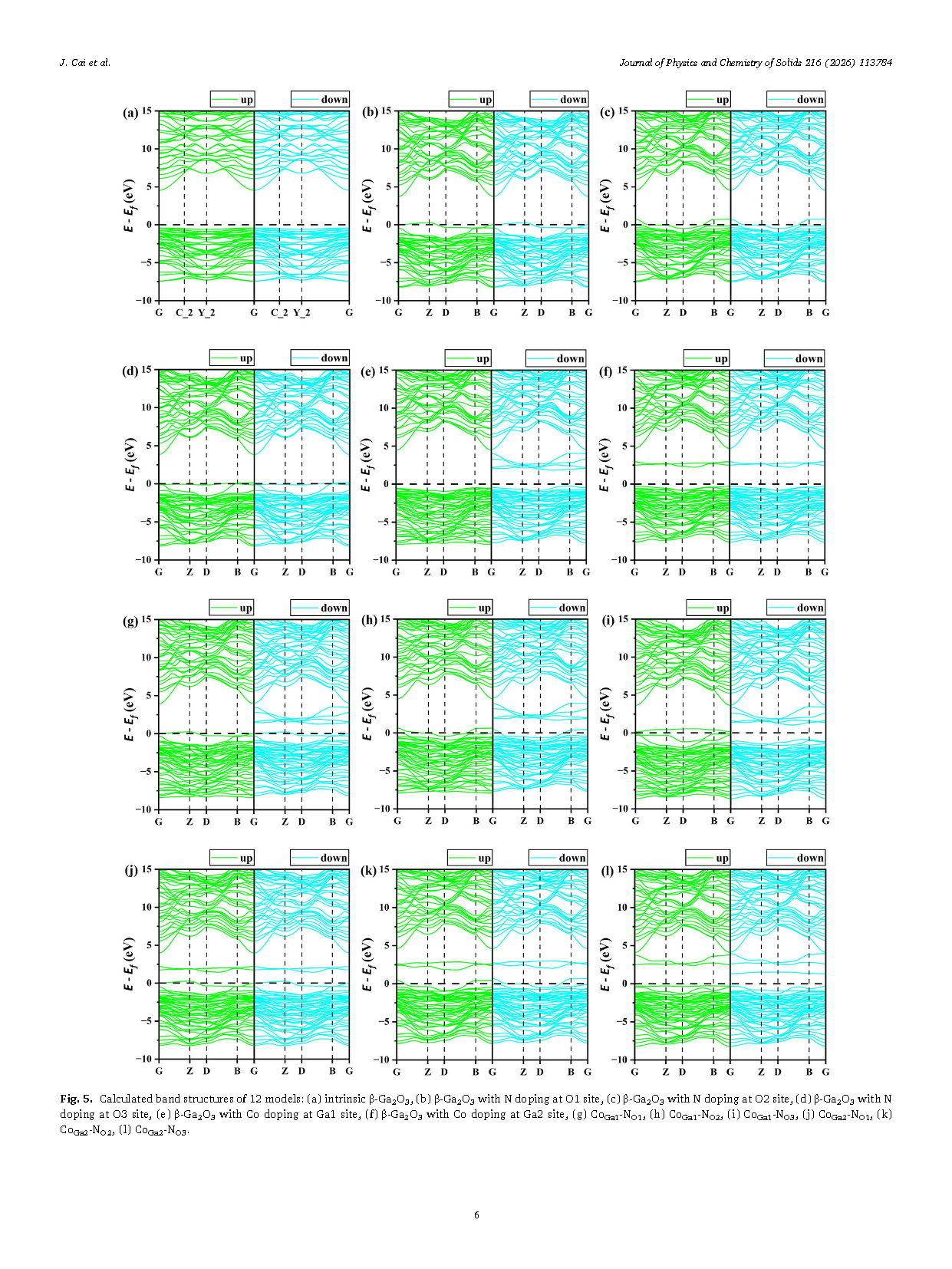
Figure 5 Calculated band structures of 12 models: (a) intrinsic β-Ga₂O₃, (b) β-Ga₂O₃ with N doping at O1 site, (c) β-Ga₂O₃ with N doping at O2 site, (d) β-Ga₂O₃ with N doping at O3 site, (e) β-Ga₂O₃ with Co doping at Ga1 site, (f) β-Ga₂O₃ with Co doping at Ga2 site, (g) CoGa1-NO1 (h) CoGa1-NO2 (i) CoGa1-NO3 (j) CoGa2-NO1 (k) CoGa2-NO2 (l) CoGa2-NO3
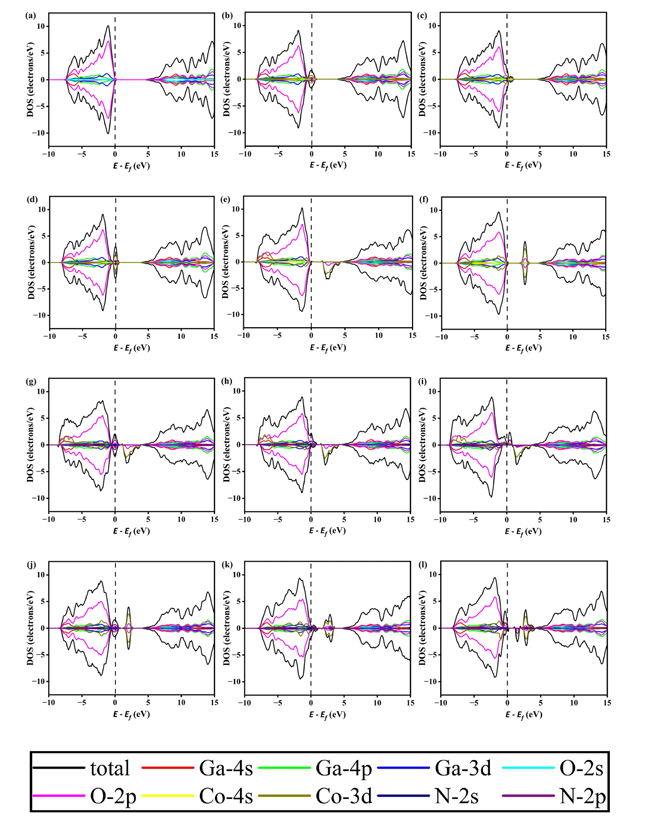
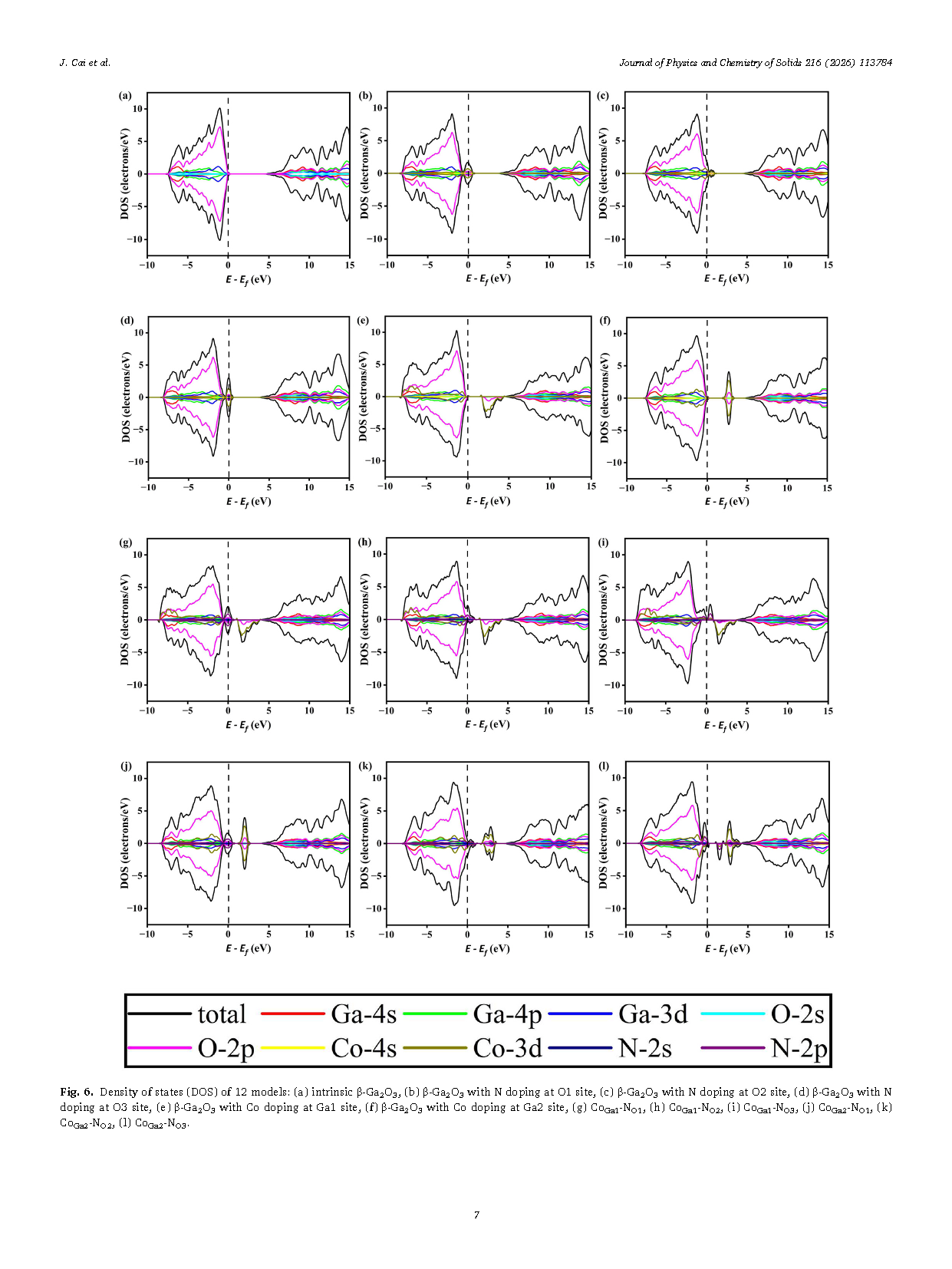
Figure 6 Density of states (DOS) of 12 models: (a) intrinsic β-Ga₂O₃, (b) β-Ga₂O₃ with N doping at O1 site, (c) β-Ga₂O₃ with N doping at O2 site, (d) β-Ga₂O₃ with N doping at O3 site, (e) β-Ga₂O₃ with Co doping at Ga1 site, (f) β-Ga₂O₃ with Co doping at Ga2 site, (g) CoGa1-NO1,(h) CoGa1-NO2,(i) CoGa1-NO3,(j) CoGa2-NO1,(k) CoGa2-NO2,(l) CoGa2-NO3
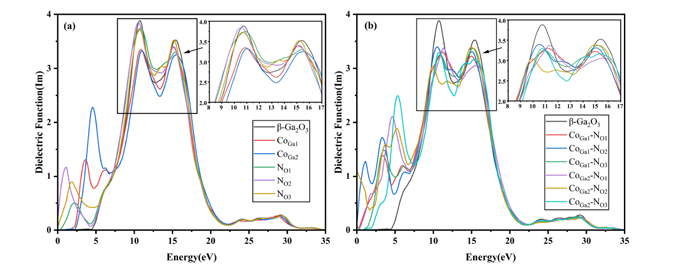
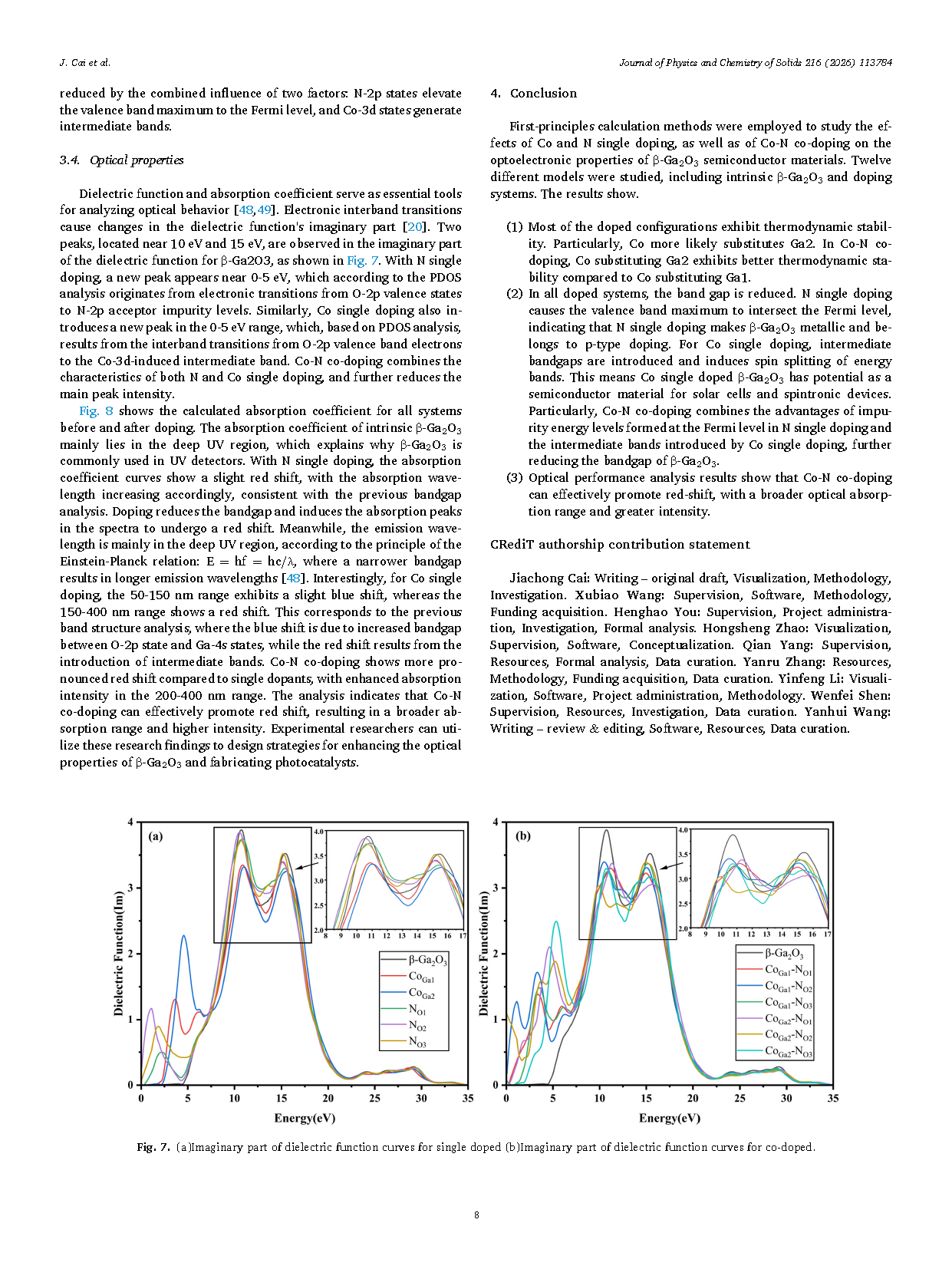
Figure 7 (a)Imaginary part of dielectric function curves for single doped (b)Imaginary part of dielectric function curves for co-doped.
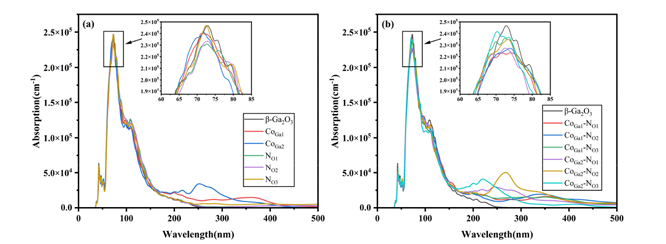
Figure 8 (a)Absorption coefficient curves for single doped (b)Absorption coefficient curves for co-doped.
DOI:
doi.org/10.1016/j.jpcs.2026.113784