Paper Sharing
【International Papers】Quantitative modeling of point defects in β-Ga₂O₃ combining hybrid functional energetics with semiconductor and processes thermodynamics
日期:2025-06-09阅读:667
Researchers from the University of Utah have published a dissertation titled "Quantitative modeling of point defects in β-Ga2O3 combining hybrid functional energetics with semiconductor and processes thermodynamics" in Physical Chemistry Chemical Physics.
Abstract
β-Gallium oxide (β-Ga2O3) is of high interest for power electronics because of its unique combination of melt growth, epitaxial growth, n-type dopability, ultrawide bandgap, and high critical field. Optimization of crystal growth processes to promote beneficial defects and suppress harmful ones requires accurate quantitative modelling of both native and impurity defects. Herein we quantitatively model defect concentrations as a function of bulk crystal growth conditions and demonstrate the necessity of including effects such as bandgap temperature dependence, chemical potentials from thermochemistry, and defect vibrational entropy in modelling based on defect formation energies computed by density functional theory (DFT) with hybrid functionals. Without these contributions, grossly-erroneous and misleading predictions arise, e.g. that n-type doping attempts would be fully compensated by Ga vacancies. Including these effects reproduces the experimental facts that melt-grown Sn-doped β-Ga2O3 crystals are conductive with small compensation while annealing the same crystals in O2 at intermediate temperatures renders them insulating. To accomplish this modeling, we developed a comprehensive modelling framework (KROGER) based on calculated defect formation energies and flexible thermodynamic conditions. These capabilities allow KROGER to capture full and partial defect equilibria amongst native defects and impurities occurring during specific semiconductor growth or fabrication processes. We use KROGER to model 873 charge-states of 259 defects involving 19 elements in conditions representing bulk crystal growth by edge-fed growth (EFG) and annealing in oxygen. Our methodology is transferrable to a wide range of materials beyond β-Ga2O3. The integration of thermodynamic and first-principles modelling of point defects provides insight into optimization of point defect populations in growth and processing.
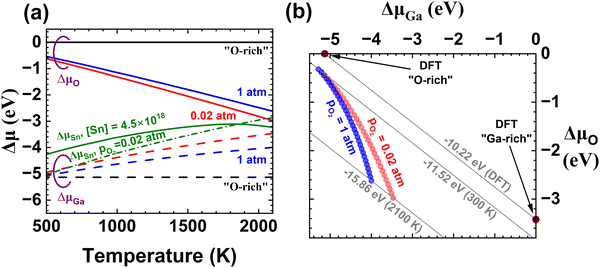
Fig. 1 (a) Temperature dependent chemical potentials for Ga, O and Sn under different thermodynamic scenarios with ΔμO solid and ΔμGa dashed: typical O- or Ga-rich (black), based on thermochemical functions from the Ga–O system for pO2 = 0.02 and 1 atm (red, blue). For Sn (green), solid denotes the case where the total [Sn] is fixed while dash-dot is derived from the SnO2 phase boundary for pO2 = 0.02 atm. (b) Plot of ΔμGa and ΔμO in Ga–O chemical potential space. At 0 or 300 K, the experimental ΔHF = ΔG° (grey diagonal lines) is within 15% of the DFT-calculated value, but as Tm is approached this error increases to 50% and the locus of conditions for which pO2 is held constant moves quite far from either O-rich or Ga-rich conditions (blue or red circles). This illustrates why using chemical potentials from thermochemistry gives a more realistic prediction for defects present.
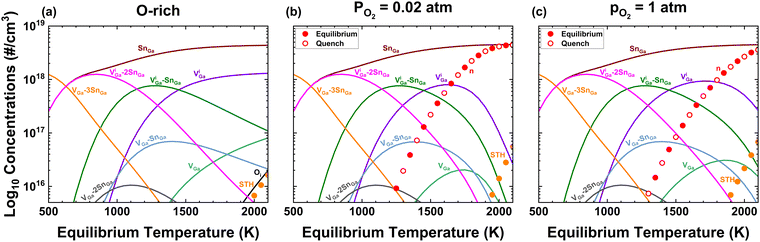
Fig. 2 Calculated concentrations of defects and holding [Sn] = 4.5 × 1018 cm−3 constant for (a) O-rich conditions holding ΔμO and ΔμGa as well as Eg constant vs. T as is typical in much existing literature. Note that the predicted carrier density is not even visible, thus samples would be predicted to be insulating. (b) For EFG crystal growth (ptot = 1 atm, pO2 = 0.02 atm, f = 0.40), and (c) O2 annealing (ptot = pO2 = 1 atm, f = 0.40). Solid circles represent equilibrium concentrations, while open circles show results for quenching from each equilibration temperature to 300 K.
DOI:
doi.org/10.1039/D4CP04817B